
Research Interest
The theoretical and computational studies in our research group are primarily aimed at exploring the mechanism of catalytic processes, and characterizing the structure, physicochemical properties and reactivity of new compounds. Additionally, we examine the properties of redox systems, the behavior of solutions, and recently we are involved in developing molecular databases as well. In mechanistic and reactivity studies, we carry out accurate quantum chemical calculations and molecular dynamics simulations, whereas we use semiempirical quantum chemical methods and machine learning approaches as well in database developments.
University students who are interested in computational chemistry are welcome in our research group.
Asymmetric catalysis
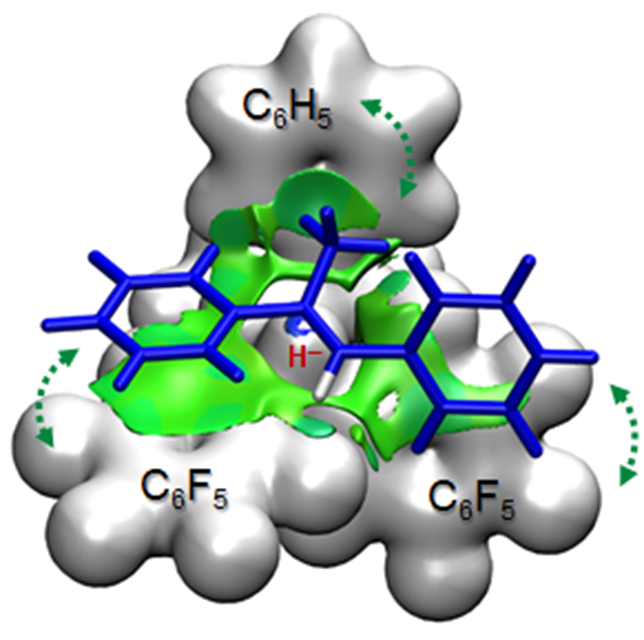
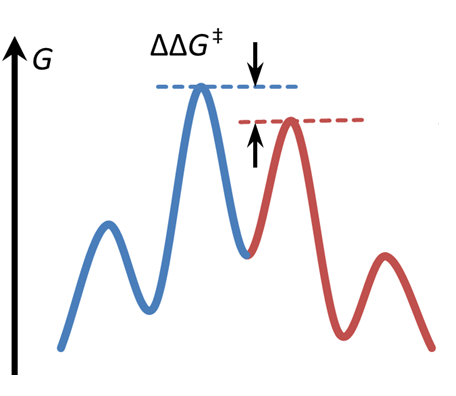
One of the major challenges in synthetic chemistry is to achieve high degree of enantioselectivity, which is generally attained by using chiral catalysts. Computational chemistry can greatly assist the synthetic developments , as computational modelling provides direct information about the structure and energetics of transition states leading to enantiomeric products. This information can be used to interpret the stereoselectivity (or the lack of stereoselectivity) observed experimentally, but computations may even give useful predictions for the design of new reactions. In this project, we examine catalytic reactions of current interest in synthetic chemistry, such as asymmetric catalytic hydrogenation, enantioselective halocyclization, and asymmetric amine catalysis.
 |
 |
Related publications:
- A. Hamza, K. Sorochkina, B. Kótai, K. Chernichenko, D. Berta, M. Bolt, M. Nieger, T. Repo, I. Pápai, Correlating electronic and catalytic properties of frustrated Lewis pairs for imine hydrogenation, ACS Catal. 10, 14290 (2020)
10.1021/acscatal.0c04263 - A. Hamza, D. Moock, C. Schlepphorst, J. Schneidewind, W. Baumann, F. Glorius, Unveiling a key catalytic pocket for the ruthenium NHC-catalysed asymmetric heteroarene hydrogenation, Chem. Sci. 13, 985 (2022)
10.1039/D1SC06409F - D. von der Heiden, F. B. Németh, M. Andreasson, D. Sethio, I. Pápai, M. Erdelyi, Are bis(pyridine)iodine(i) complexes applicable for asymmetric halogenation?, Org. Biomol. Chem. 19, 8307 (2021)
10.1039/D1OB01532J
Photochemistry and excited state reactivity
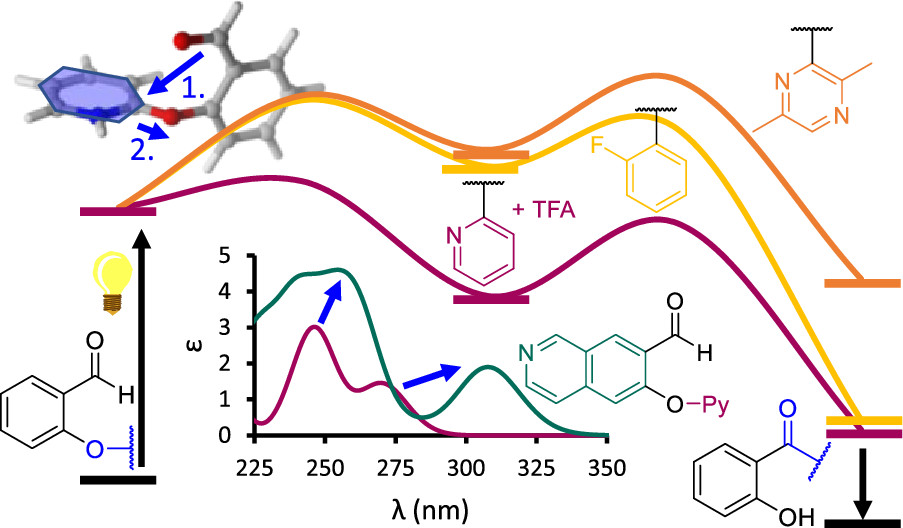
One of the most actively studied areas of chemistry today is the use of solar energy to create or break chemical bonds. The key factor is to understand the reaction mechanism. For this purpose, computational chemistry is indispensable as it allows to describe the change in electronic structure upon excitation by light, which is either too expensive or impossible to observe directly via experiments. Computations can also accurately predict photophysical properties such as UV-vis spectra, solvatochromic behaviors, charge transfer characters or excited state redox potentials of molecules not yet synthesized. With this knowledge, we can identify promising photoactive molecules or exclude inactive ones to make synthesis more efficient. Our group is engaged in the study of excited state reaction mechanisms, which includes the area of photocatalysis. We also contribute to the design of novel fluorescent probe molecules. In method development, we design approaches for UV-vis spectrum prediction.
 |
Related publications:
- A. Adamoczky, T. Nagy, P. P. Fehér, V. Pardi-Tóth, Á. Kuki, L. Nagy, M. Zsuga, S. Kéki, Isocyanonaphthol Derivatives: Excited-State Proton Transfer and Solvatochromic Properties, Int. J. Mol. Sci. 23, 7250 (2022)
10.3390/ijms23137250 - P. P. Fehér, Á. Madarász, A. Stirling, Multiscale Modeling of Electronic Spectra Including Nuclear Quantum Effects, J. Chem. Theory Comput. 17, 6340 (2021)
10.1021/acs.jctc.1c00531 - P. P. Fehér, Density Functional Theory Evaluation of a Photoinduced Intramolecular Aryl Ether Rearrangement, J. Org. Chem. 86, 2706 (2021)
10.1021/acs.joc.0c02706
Nuclear quantum effects
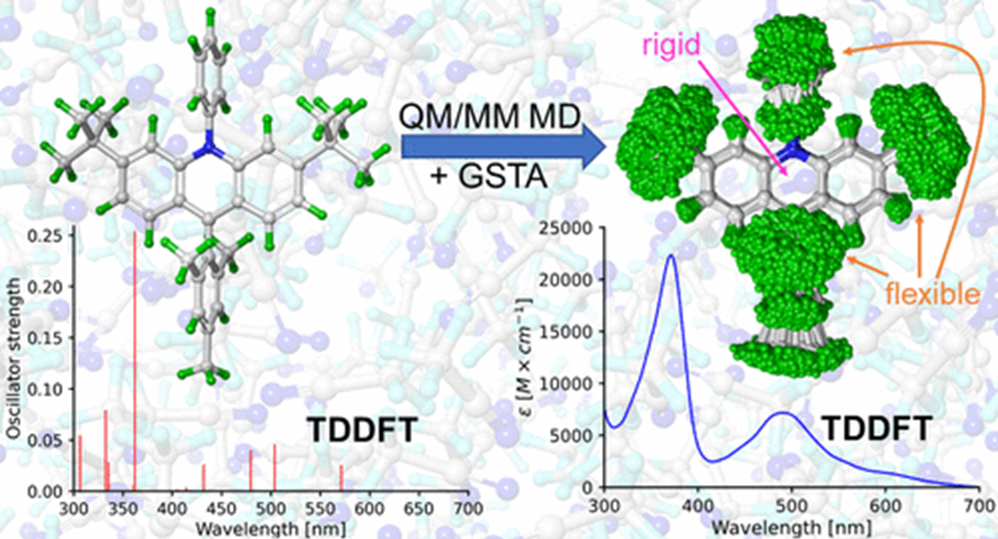
The atomic level description of chemical reactions often requires the consideration of nuclear quantum effects. In this project we intend to assess the importance of these effects via theoretical chemical methods. We wish to introduce further developments to our GSTA procedure and assess its applicability. Based on available experimental data and path-integral molecular dynamics simulations, we develop and test new methods that can be applied to characterize various properties of compounds and solvents used in chemical synthesis and catalysis. We primarily focus on thermodynamic properties, but we analyze the structural, kinetic, and spectroscopic properties as well.
 |
Related publications:
- D. Berta, D. Ferenc, I. Bakó, A. Madarász, Nuclear quantum effects from the analysis of smoothed trajectories: Pilot study for water, J. Chem. Theory Comput. 16, 3316 (2020)
10.1021/acs.jctc.9b00703 - I. Bakó, Á. Madarász, L. Pusztai, Nuclear quantum effects: Their relevance in neutron diffraction studies of liquid water, J. Mol. Liq. 325, 115192 (2021)
10.1016/j.molliq.2020.115192 - P. Fehér, Á. Madarász, A. Stirling, Multiscale Modeling of Electronic Spectra Including Nuclear Quantum Effects, J. Chem. Theory Comput. 17, 6340 (2021)
10.1021/acs.jctc.1c00531
Hydration of macromolecules
When it comes to reactions taking place in protic solvents, the reacting molecules form complex H-bonding networks with the environment. To characterize the topology of these H-bonded structures, we apply statistical physics and network theory methods. Using various parameters obtained from quantum chemical calculations, we investigate the strength and the nature of the H-bonds formed between the solute molecule and the surrounding water molecules. This project focuses on hydrated macromolecules (sugars, proteins, DNA, cyclodextrins) and we examine the effect hydration on the native structures and the dynamic properties of these species. We explore the influence of the nuclear quantum effects on structural, dynamic and thermodynamic properties using the GSTA method.
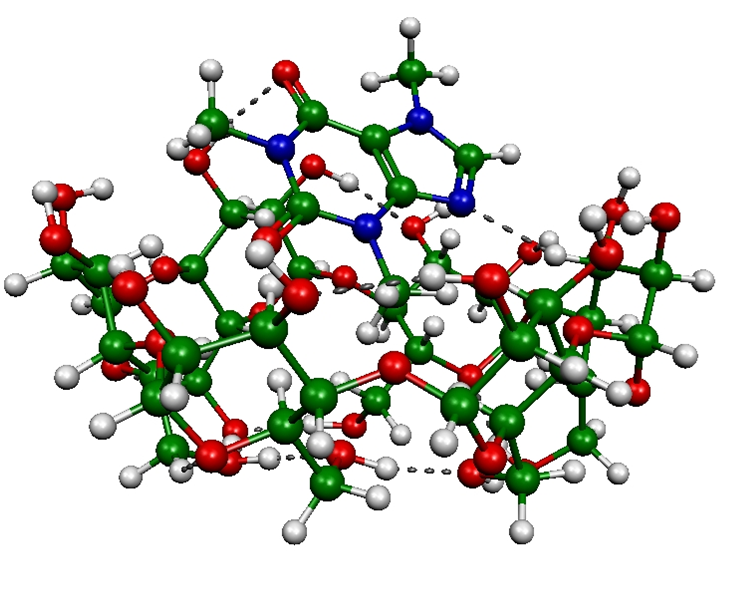 |
Related publications:
- Sz. Pothoczki, I. Pethes, L. Pusztai, L. Temleitner, K. Ohara, I. Bakó, Properties of Hydrogen-Bonded Networks in Ethanol–Water Liquid Mixtures as a Function of Temperature: Diffraction Experiments and Computer Simulations, J. Phys. Chem. B 125, 6272 (2021)
10.1021/acs.jpcb.1c03122 - A. Pethes, I. Bakó, L. Pusztai, Chloride ions as integral parts of hydrogen bonded networks in aqueous salt solutions: the appearance of solvent separated anion pairs, Phys. Chem. Chem. Phys. 22, 11038 (2020)
10.1039/D0CP01806F
Computational screening of redox active organic compounds:
The participants of the EU Horizon 2020 CompBat project aimed at identifying new prospective organic molecules for next generation redox flow batteries. In this project, our research group focuses on the development of computational procedures that enable large-scale virtual screening of molecules. We use various cheminformatics tools and quantum chemical methods to construct extended molecular databases, and we apply machine learning approaches to assist the screening. Our database currently includes the structures and standard reduction potentials of over 10.000 organic molecules. The related stability studies and the results of deep learning applications may provide useful guidance for synthetic developments and electrochemical studies.
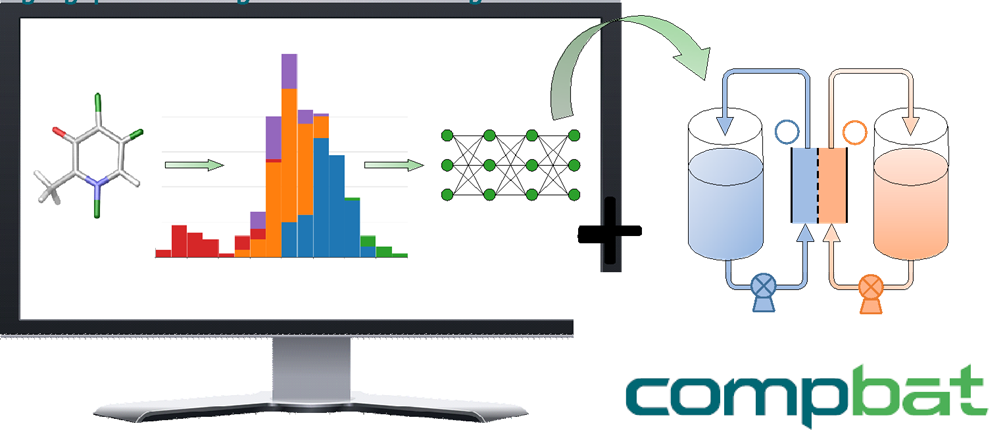 |
Further Information:
- The website of the CompBat project
- Short movie introducing our research group (English subtitles are available)
Collaborations
- University of Jyväskylä, Finland
- University of Helsinki, Finland
- University of Bari, Italy
- Universitat Autonoma de Barcelona, Spain
- University of Girona, Spain
- Angstrom Laboratory, Uppsala University, Sweden
Education
- PhD supervision (Hevesy György PhD School of Chemistry, Eötvös Loránd University)
- Bachelor and master thesis supervision (ELTE TTK, BME)
Leader
Imre Pápai