Kutatócsoport-vezető
Szüts Dávid DSc
Igazgatóhelyettes, Molekuláris Élettudományi Intézet
A kutatócsoport része az Ektópiás Meszesedés Laboratórium, témavezető: Szeri Flóra PhD
Általános kutatási terület
A genomiális DNS állomány állandóan károsító hatásoknak van kitéve, melyek környezeti vagy endogén eredetűek lehetnek. Bár az élő sejtek rendelkeznek DNS-javító mechanizmusokkal, osztódásra készülő sejtekben elkerülhetetlen, hogy a DNS replikáció sérült DNS templát szakaszokba ütközzön. Az elakadt replikáció a sejtciklus leállásához és sejthalálhoz vezethet, ezért több olyan molekuláris mechanizmus is létezik, amely segíti a replikáció továbbhaladását sérült DNS-szakaszokon. A sérült DNS sikeres replikációja lehetővé teszi a sejt túlélését, ugyanakkor a folyamat a mutációk fő forrása. A szomatikus sejtekben bekövetkező mutagenezis a daganatképződés fő okozója, és a genomi instabilitás a rák egyik általános jellemzője. Kutatócsoportunk a sérült DNS replikációjának mechanizmusát és következményeit vizsgálja, különös tekintettel a mutagenikus folyamatok szerepére a rák kialakulásában és kezelésében.
Főbb kutatási témák
A DNS-hibaelkerülő útvonalak szabályozása
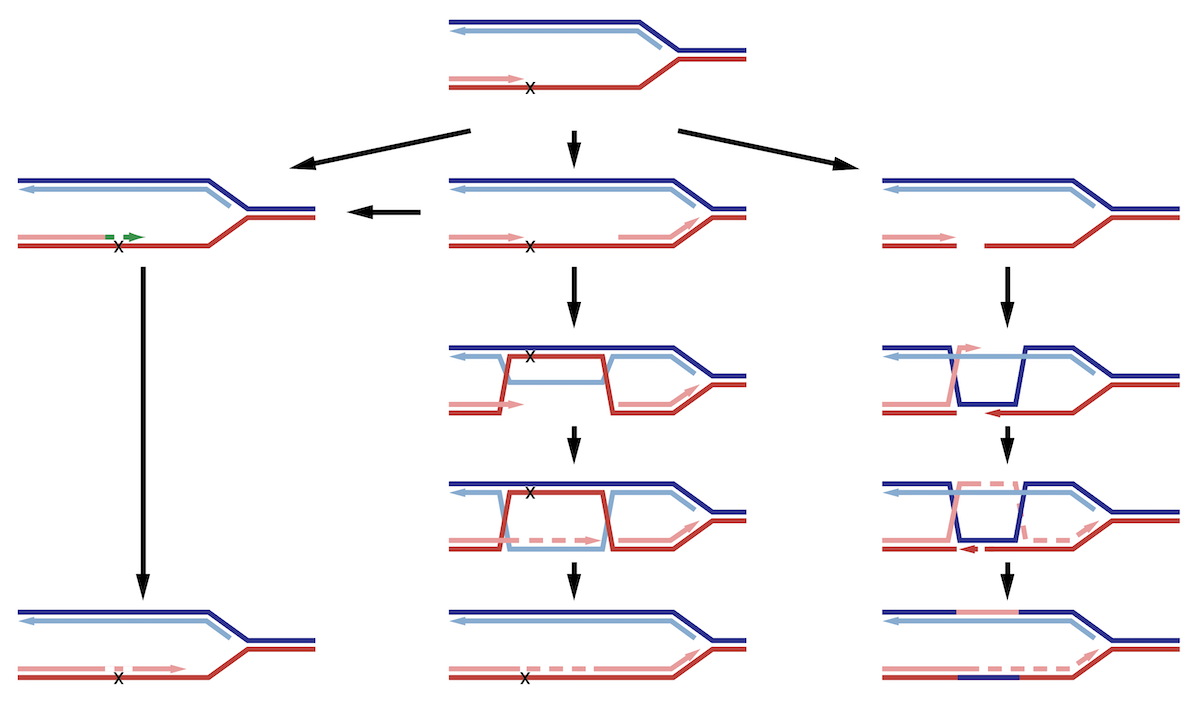
Legalább háromféle útvonal létezik az DNS lézióknál elakadt replikáció továbbsegítésére (2. ábra). A transzléziós szintézis speciális polimerázokat segítségével halad át a sérült templáton, és hajlamos pont mutációkat előidézni az új szálban. A templátváltás és a homológ rekombináció mechanizmusai viszont egyaránt lehetővé teszik a lézió elkerülését, a testvérkromatída új szálát használva templátként a sérült szakasznál. Vizsgáljuk a megfelelő útvonal kiválasztását szabályozó mechanizmusokat, melyek befolyásolják a genomban kialakuló mutációk típusát és gyakoriságát.
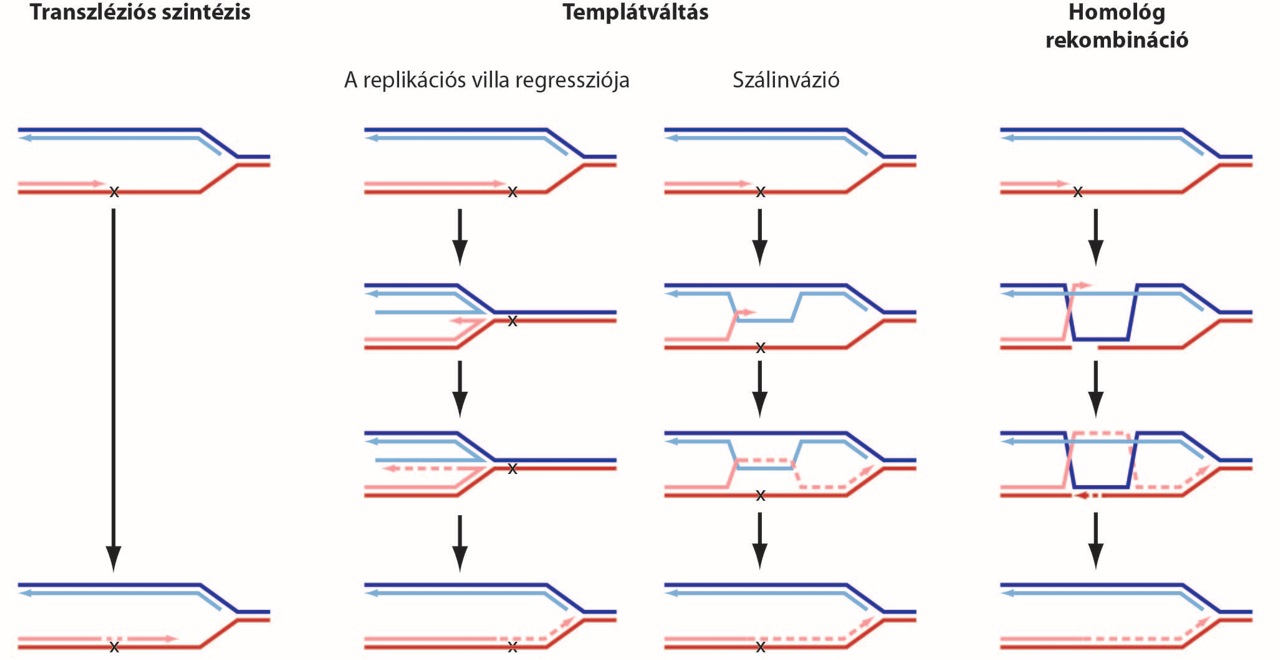
Transzléziós szintézis
Kutatásaink során vizsgáljuk a folyamatban résztvevő fehérjék és kovalens fehérjemódosítások szerepét a megfelelő polimeráz kiválasztásában és a transzléziós szintézis időbeli szabályozásában. Ezen felül vizsgáljuk, hogy a keletkezett mutációk spektruma hogyan függ a léziók típusától, a reakcióban részt vevő polimeráztól és a szabályozó folyamatoktól.
A rákra jellemző mutagenikus folyamatok modellezése
Génkiütött sejtvonalak létrehozásával tumormodelleket állítunk elő, melyeken nagy áteresztőképességű, teljes genomra kiterjedő mérésekkel vizsgáljuk a tumorsejtek genomiális instabilitásának okait és következményeit. Elsősorban a rosszul működő DNS-javító folyamatok következtében, illetve kemoterápiás kezelések hatására fellépő mutagenezist kutatjuk.
Genetika a DT40 sejtvonalban
Kutatásunk nagymértékben támaszkodik a csirke DT40 sejtvonalból előállított génkiütött és egyéb mutáns sejtvonalakon végzett kísérletekre. A DT40 limfoblasztóma sejtvonal a gerinces sejtkultúra-rendszerek között kimagaslóan alkalmas a célzott génmódosítások előállítására. A DT40 sejtek alkalmazásának komoly hagyománya van a DNS-javítás területén, és a folyamatokban szerepet játszó fehérjék konzerváltsága miatt az eredmények az emberi sejtekben működő mechanizmusok megértéséhez teljes mértékben relevánsak. A gyorsan osztódó DT40 sejtek különösen jól alkalmazhatóak a sérült DNS replikációjának genetikai vizsgálatára. Megszekvenáltuk és jellemeztük a DT40 sejtvonal genomját, mely elérhető letöltés és BLAST keresések céljából: http://dt40.enzim.ttk.mta.hu
Genomi elemzések és módszerfejlesztés
Az újgenerációs DNS-szekvenálás lehetővé teszi a mutagenezis folyamatának genoszintű elemzését. Kutatásainkhoz genomszekvenálást használunk a DNS-javító folyamatok mutagenezisre gyakorolt befolyásának vizsgálatára. Ehhez teljes genomszekvencia-adatokon alapuló mutáció-detektálási módszereket is fejlesztünk bioinformatikai csoportokkal együttműködve.
Fontosabb publikációk
Lózsa R, Németh E, Gervai JZ, Márkus BG, Kollarics S, Gyüre Z, Tóth J, Simon F, Szüts D. (2023). DNA mismatch repair protects the genome from oxygen-induced replicative mutagenesis. Nucleic Acids Res. 10.1093/nar/gkad775
Gyüre Z, Póti Á, Németh E, Szikriszt B, Lózsa R, Krawczyk M, Richardson AL, Szüts D. (2023). Spontaneous mutagenesis in human cells is controlled by REV1-Polymerase ζ and PRIMPOL. Cell Rep. 42, 112887
Szüts D. (2022). A fresh look at somatic mutations in cancer. Science 376, 351-352.
Chen D, Gervai JZ, Póti Á, Németh E, Szeltner Z, Szikriszt B, Gyüre Z, Zámborszky J, Ceccon M, d’Adda di Fagagna F, Szallasi Z, Richardson AL, Szüts D. (2022). BRCA1 deficiency specific base substitution mutagenesis is dependent on translesion synthesis and regulated by 53BP1. Nat Commun. 13, 226.
Póti Á, Szikriszt B, Gervai JZ, Chen D, Szüts D. (2022). Characterisation of the spectrum and genetic dependence of collateral mutations induced by translesion DNA synthesis. PLoS Genet. 18, e1010051.
Szikriszt B, Póti Á, Németh E, Kanu N, Swanton C, Szüts D. (2021). A comparative analysis of the mutagenicity of platinum-containing chemotherapeutic agents reveals direct and indirect mutagenic mechanisms. Mutagenesis 36, 75-86.
Németh E, Lovrics A, Gervai JZ, Seki M, Rospo G, Bardelli A, Szüts D. (2020). Two main mutational processes operate in the absence of DNA mismatch repair. DNA Repair (Amst). 89, 102827.
Póti Á, Gyergyák H, Németh E, Rusz O, Tóth S, Kovácsházi C, Chen D, Szikriszt B, Spisák S, Takeda S, Szakács G, Szallasi Z, Richardson AL, Szüts D. (2019). Correlation of homologous recombination deficiency induced mutational signatures with sensitivity to PARP inhibitors and cytotoxic agents. Genome Biol. 20, 240.
Németh E, Krzystanek M, Reiniger L, Ribli D, Pipek O, Sztupinszki Z, Glasz T, Csabai I, Moldvay J, Szallasi Z, Szüts D. (2019). The genomic imprint of cancer therapies helps timing the formation of metastases. Int J Cancer 145, 694-704.
Póti Á, Berta K, Xiao Y, Pipek O, Klus GT, Ried T, Csabai I, Wilcoxen K, Mikule K, Szallasi Z, Szüts D. (2018). Long-term treatment with the PARP inhibitor niraparib does not increase the mutation load in cell line models and tumour xenografts. Br J Cancer 119, 1392-1400.
Gervai JZ, Gálicza J, Szeltner Z, Zámborszky J, Szüts D. (2017). A genetic study based on PCNA-ubiquitin fusions reveals no requirement for PCNA polyubiquitylation in DNA damage tolerance. DNA Repair (Amst). 54, 46-54.
Zámborszky J, Szikriszt B, Gervai J, Pipek O, Póti Á, Ribli D, Krzystanek M, Szalai-Gindl JM, Swanton C, Szallasi Z, Csabai I, Richardson AL, Szüts D. (2017). Loss of BRCA1 or BRCA2 markedly increases the rate of base substitution mutagenesis and has distinct effects on genomic deletions. Oncogene 36, 746-755.
Szikriszt B, Póti Á, Pipek O, Krzystanek M, Kanu N, Molnár J, Ribli D, Szeltner Z, Tusnády GE, Csabai I, Szállási Z, Swanton C, Szüts D. (2016). A comprehensive curvey of the mutagenic impact of common cancer cytotoxics. Genome Biol. 17, 99.
Együttműködések
Nemzetközi együttműködések
- IFOM ETS – The AIRC Institute of Molecular Oncology, Milano, Italy
- The Francis Crick Institute, London, UK
- MRC Laboratory of Molecular Biology, Cambridge, UK
- Johns Hopkins University, Baltimore, USA
- Washington University, St. Louis, USA
- Boston Children’s Hospital, Boston, USA
- Danish Cancer Society Research Center, Copenhagen, Denmark
Magyarországi együttműködések
- HUN-REN TTK Szerkezetkutató központ
- HUN-REN SZBK Genetikai Intézet
- HUN-REN Rényi Alfréd Matematikai Kutatóintézet
- ELTE Komplex Rendszerek Fizikája Tanszék
- Budapesti Műszaki Egyetem, Fizika Tanszék
- Debreceni Egyetem, Gyógyszertechnológiai Tanszék
- Semmelweis Egyetem Patológiai és Kísérleti Rákkutató Intézet

A kutatócsoport tagjai
Genomstabilitás kutatócsoport
- Szüts Dávid DSc (publikációk)
- Szeltner Zoltán PhD (publikációk)
- Szikriszt Bernadett PhD (publikációk)
- Németh Eszter PhD (publikációk)
- Póti Ádám PhD (publikációk)
- Michał Krawczyk PhD
- Szabó Bendegúz, PhD hallgató
- Martinek Regina, PhD hallgató
- Engel Botond, PhD hallgató
Ektópiás meszesedés laboratórium (honlap)
- Szeri Flóra PhD (publikációk)
- Virgil Tamatey, PhD hallgató
- Várhegyi Martin, PhD hallgató
Végzett PhD hallgatók
- 2018 Gervai Judit
- 2022 Póti Ádám
- 2023 Gyüre Zsolt